
医薬品の品質保証や品質管理の現場において、膨大な文書作成やデータの整合性確認に追われ、頭を悩ませてはいませんか?
近年、製薬業界では「データインテグリティ(データの完全性)」への対応が厳格化されており、現場の負担は増すばかりです。しかし、要件を正しく理解し、適切なツールを活用することで、リスクを減らしながら業務を効率化することが可能です。
この記事では、データインテグリティの基本からALCOA原則、そして最新のAI技術を用いた解決策までをわかりやすく解説します。
この記事を読み終える頃には、データインテグリティ対応への不安が解消され、実務の効率化に向けた具体的な一歩が見えてくるはずです。
目次
データインテグリティとは?製薬業界で求められる背景
まず、「データインテグリティ(Data Integrity)」という言葉の意味と、なぜ今、製薬業界でこれほどまでに重要視されているのかを解説します。
データインテグリティの定義
データインテグリティとは、直訳すると「データの完全性」や「データの整合性」を意味します。製薬業界においては、データのライフサイクル全体を通じて、データが「完全で、一貫性があり、正確であること」を指します。
具体的には、医薬品の製造や品質管理の過程で生成されたデータが、改ざんや欠落なく、事実そのものであることを保証する概念です。
注目される背景と規制の強化
近年、国内外でデータ改ざんや不適切なデータ管理による品質問題が相次いで報告されました。これを受け、PMDA(独立行政法人 医薬品医療機器総合機構)やPIC/S(医薬品査察協定・医薬品査察共同スキーム)といった規制当局は、データ管理に対する監視を強めています。
特にPMDAは「医薬品の製造管理及び品質管理の基準(GMP)ガイドライン」等の各種ガイドラインの中で、データの信頼性確保を強く求めています。適切なデータインテグリティ対応ができていない場合、規制当局からの指摘事項となり、最悪の場合、業務停止命令や社会的信用の失墜につながるリスクがあります。
GMPにおけるデータインテグリティと「ALCOA原則」

GMP(Good Manufacturing Practice:医薬品の製造管理及び品質管理の基準)の現場でデータインテグリティを確保するために、必ず押さえておくべき基準が「ALCOA(アルコア)原則」です。
ALCOA(アルコア)原則とは
ALCOA原則とは、データの信頼性を担保するために満たすべき5つの要件の頭文字をとったものです。
- A:Attributable(帰属性)
誰が、いつその作業を行ったかが明確であること。作業者の署名やタイムスタンプなどが該当します。 - L:Legible(判読性)
データが読みやすく、長期にわたって保存・参照可能であること。手書きの記録が汚れて読めないといった状態はNGです。 - C:Contemporaneous(同時性)
作業が行われたその時に記録されていること。後から記憶を頼りに記録する「バックデート」は認められません。 - O:Original(原本性)
最初に記録されたデータ、または認定された写しであること。 - A:Accurate(正確性)
データが事実通りであり、誤差や修正がある場合はその履歴が残っていること。
これらの原則は、紙の記録だけでなく、電子データにおいても同様に適用されます。
さらに進化した「ALCOA+(アルコアプラス)」
近年では、上記の5つに加えて、以下の4つの要素を加えた「ALCOA+(CCEA)」の遵守も求められています。
- Complete(完全性): データが欠落なくすべて揃っていること。
- Consistent(一貫性): データの日時や順序に矛盾がないこと。
- Enduring(耐久性): データが記録媒体に長期間保存され、消失しないこと。
- Available(利用可能性): 必要なときにいつでもデータにアクセスできること。
これらを徹底することは、製品の品質を保証し、患者様の安全を守るための「義務」であるといえます。
FDAから実際に指摘された3つのデータインテグリティ違反事例(2024年)
FDA(米国食品医薬品局)は、データインテグリティ違反を毎年多くの製薬企業に指摘しています。複数の調査によれば、近年のFDA Warning Letter(警告書)の60〜80%にデータインテグリティに関する指摘が含まれているとされています。
ここでは、2024年に実際にFDAが発出した警告書の中から、典型的な3つの違反事例を取り上げます。いずれもFDA公式サイトで全文が公開されている一次情報です。
事例1:Sichuan Deebio Pharmaceutical(中国)|微生物試験プレートの非同時記録
2024年2月5日、FDAのCDER(医薬品評価研究センター)は、中国・四川省のSichuan Deebio Pharmaceutical社(API=原薬製造企業)に対して警告書を発出しました。
2023年9月に実施された査察で、QC(品質管理)の微生物試験室における重大な逸脱が確認されたためです。FDAは「査察官は、多数の微生物試験プレートが同時に読み取られ、記録されていないことを確認した」と指摘し、「実験室記録での非同時記録は、当社の試験記録の妥当性とインテグリティに対する懸念を生じさせる」と記載しました。
これは、ALCOA+原則の「Contemporaneous(同時性)」違反の典型例です。
事例2:Optikem International(米国)|文書管理とバッチ記録の不備
2024年6月20日、FDAは米国Optikem International社に対して警告書を発出しました。同社の2024年2月の回答について、FDAは「適切な文書管理とデータインテグリティを確保するための計画について、十分な詳細を提供していない」「バッチ記録の文書化、データ管理、無菌処理オペレーターの適格性確認手順の変更が回答に欠けている」と指摘しました。特に重要なのは、FDAが「試験結果の完全かつ正確な記録の作成または維持に失敗した場合、データの信頼性が根本的に損なわれる」と明記している点です。これはALCOA+原則の「Complete(完全性)」「Accurate(正確性)」の違反事例です。
事例3:Laboratorio Magnachem International(ドミニカ共和国)|HPLCシステムの電子記録管理不備
2024年6月18日、FDAはドミニカ共和国の医薬品メーカーLaboratorio Magnachem International社に対して警告書を発出しました。2023年11月の査察を受けたもので、HPLCシステムと電子記録管理におけるデータインテグリティの違反が指摘されました。
FDAは、同社が「CGMP電子データに対する厳格な管理を維持できなかった」「追加・削除・修正が承認され、適切に文書化されていることを保証できなかった」「患者保護のための暫定措置を欠いていた」と指摘。Broncochem製品を含む製品バッチが、データインテグリティのギャップに関連する安定性試験の不備によりリコール対象となっています。
参照: FDA「Sichuan Deebio Pharmaceutical Co. Ltd Warning Letter」「Optikem International Inc. Warning Letter」「Laboratorio Magnachem International Warning Letter」
データインテグリティへの対応のポイントと実務

では、具体的にどのように対応を進めればよいのでしょうか。
GMPの現場においてデータインテグリティを確保するためには、単にシステムを導入するだけでなく、「ガバナンス」「技術」「手順」の3つの側面から包括的に対策を講じる必要があります。
① ガバナンス:経営層のコミットメントと企業風土
データインテグリティ対応の第一歩は、現場の担当者ではなく、経営層(シニアマネジメント)の姿勢にあります。
- 品質文化(Quality Culture)の醸成: 「悪い報告をしても責められない」「データの隠蔽や改ざんが許されない」というオープンな企業風土を作ることが不可欠です。現場が納期やコストのプレッシャーから不正に手を染めないよう、経営層がリソース(人員・時間)を適切に配分する必要があります。
- 教育・トレーニングの徹底: 全従業員に対し、データインテグリティの重要性と、不正が患者様の命に関わるリスクであることを定期的に教育します。
② 技術的対策(Technical Controls):システムによる制御
意図しないミスや不正を防ぐため、コンピュータ化システムには以下の機能要件が求められます。
- アクセス管理の厳格化: ユーザーごとに固有のIDとパスワードを発行し、共有(使い回し)を禁止します。また、管理者(Administrator)権限と、分析を行う担当者(User)の権限を明確に分離し、担当者が自分のデータを削除・変更できない設定にします。
- 監査証跡(Audit Trail)のレビュー: 「誰が、いつ、何を、なぜ変更したか」を自動記録する監査証跡機能を有効にします。重要なのは記録することだけでなく、その履歴を定期的にレビュー(点検)し、不審な操作がないかを確認するプロセスを構築することです。
③ 手順的対策(Procedural Controls):人の行動とルールの徹底
システムで制御しきれない部分(紙の記録や手作業)は、SOP(標準作業手順書)でカバーします。
- ブランクフォーム(空欄用紙)の管理: 「良い結果が出るまで何度も試験を行い、悪い結果の記録を捨てる」といった行為を防ぐため、発行枚数を管理した番号付きの記録用紙(ブランクフォーム)を使用し、書き損じも含めて全て回収・保管します。
- 生データの定義と保存: 何をもって「原本(オリジナル)」とするかを定義します。電子天秤の印字レシートや、チャート図などの生データは、劣化しないよう適切に保存する必要があります。
④ リスクマネジメント:データライフサイクルの把握
すべてのデータに同じ労力をかけるのではなく、リスクベースアプローチを採用します。
- データライフサイクルの特定: データが生成されてから、処理、保存、アーカイブ、廃棄されるまでの流れ(ライフサイクル)を可視化します。
- リスク評価(Risk Assessment): データの重要度や、改ざん・ミスの起きやすさを評価し、リスクが高いプロセスに優先的に対策リソースを投入します。
現場が抱える「文書作成」の負担と課題
これらの対策(SOPの整備、リスク評価書の作成、監査証跡のレビュー記録など)をすべて実行するには、膨大な文書作成業務やチェック業務が発生します。 「手順書と実際の記録に齟齬がないか」「法規制の最新版に対応できているか」といった確認作業は、すべて人の手で行うには限界があります。薬機法が年々厳格化していく中、多くの品質保証担当者が本来注力すべき品質改善業務よりも、書類の整合性チェックに多くの時間を奪われているのが現状です。
【自社チェック用】ALCOA+対応チェックリスト
自社のデータインテグリティ対応状況を点検する際に活用できるチェックリストです。紙記録・電子記録の両方の観点から、ALCOA+原則の主要要件をカバーしています。査察対応のセルフチェックとしてもご活用ください。
| カテゴリ | チェック項目 |
| Attributable(帰属性) | □ 紙記録:すべての記入欄に作業者の署名・記入日が記録されているか □ 電子記録:ユーザーIDが個人に紐づき、共有アカウントの使用がないか □ 監査証跡(誰が/いつ/何を/なぜ変更したか)が自動記録されているか |
| Legible(判読性) | □ 紙記録:消えにくいインクが使われ、訂正は二重線+訂正者の押印・日付が記載されているか □ 電子記録:保管期間中も読み取り可能なフォーマットで保存されているか □ 廃止文書を判別できる仕組み(廃版印など)があるか |
| Contemporaneous(同時性) | □ 作業発生時にリアルタイムで記録されているか(バックデートの禁止) □ 電子システムの時刻同期(NTP等)が行われているか □ 記録のタイムスタンプは作業実施時刻と一致しているか |
| Original(原本性) | □ 原本データの改変不可な保管が行われているか□ 写しを使う場合はCertified True Copyとして管理されているか □ 電子データの完全な再現が可能か |
| Accurate(正確性) | □ 測定機器のキャリブレーション・点検記録が保管されているか □ ダブルチェック・QAレビューのプロセスが定着しているか □ 入力誤りを検出・是正する仕組みがあるか |
| Complete(完全性) | □ 失敗試験・逸脱記録も含め、すべてのデータが破棄されず保管されているか□ 試験未実施・未記録の項目が残っていないか□ 例外データ(再試験・除外データ)の理由が記録されているか |
| Consistent(一貫性) | □ 時系列・関連項目間でデータに矛盾がないか □ 関連文書(試験記録/製造記録/逸脱記録)のデータが整合しているか |
| Enduring(耐久性) | □ 保管期間中(GMP省令で定める期間)の劣化・消失リスクが管理されているか □ 電子記録の定期バックアップが実施されているか □ 記録媒体の耐用年数が管理されているか |
| Available(利用可能性) | □ 必要時に速やかにデータへアクセスできる仕組みがあるか □ 検索性が確保されているか □ 適切なアクセス権限管理が行われているか |
AI活用で変わる品質保証 | QAI Generatorのご紹介
こうした課題を解決し、データインテグリティの確保と業務効率化を両立させる手段として、AI(人工知能)の活用が注目されています。
AIによる自動化でヒューマンエラーを削減
最新のAI技術を活用することで、これまで人が行っていた文書の作成やチェック作業を大幅に効率化できます。AIは疲れを知らず、膨大なデータの中から整合性の不備や記載ミスを瞬時に検出することが可能です。これにより、ヒューマンエラーによるデータインテグリティ欠如のリスクを最小限に抑えることができます。
【実例】万協製薬×EQUES|QAI GeneratorでGMP文書作成時間を5割・レビュー時間を7割以上削減
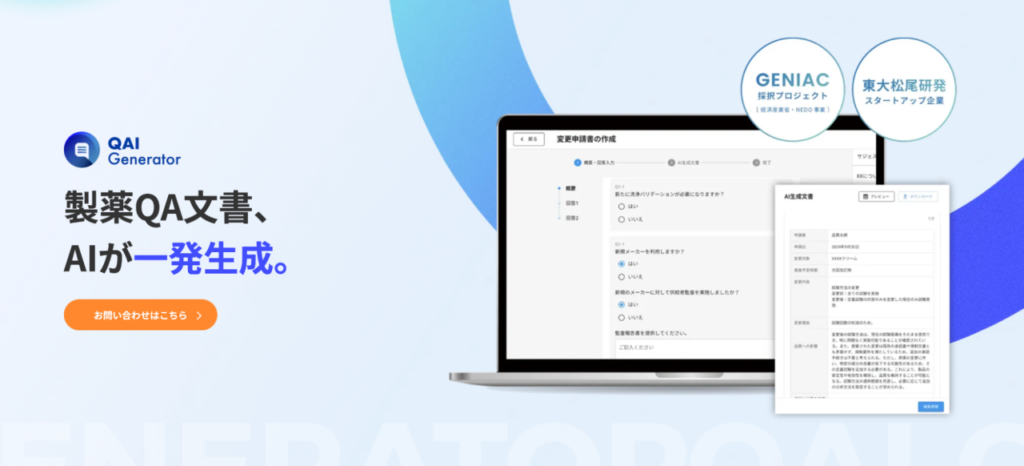
データインテグリティを担保しつつ、現場の業務負荷を減らすには、AIの活用が有力な解決策となります。ここでは、外用薬の受託製造で知られる万協製薬株式会社(三重県)が、EQUESの「QAI Generator」を導入したことで、GMP文書業務を大幅に効率化した実例を紹介します。
万協製薬では、変更申請書・逸脱報告書をはじめとするGMP文書の作成・レビューに、現場担当者の多くの時間が割かれていました。文書をゼロから作成する作業に加え、ALCOA+原則に沿ったレビューを上長が行うため、差し戻しと修正の繰り返しが大きな負担となっていたのです。
EQUESのQAI Generatorを導入した結果、文書カテゴリに応じた質問に単語や簡単な文章で回答するだけで、GMP文書の草案がAIによって自動生成されるようになりました。これにより、文書作成時間は約50%、上長によるレビュー時間は70%以上削減されています。
QAI Generatorの特徴
QAI Generatorの特長は、汎用の生成AIでは対応が難しい製薬規制への適合性を、文書種類別のアルゴリズムによって実現している点です。これにより、ALCOA+原則の「Accurate(正確性)」「Complete(完全性)」を担保しながら、GMP文書業務の効率化を進めることが可能になります。2026年3月のアップデートでは、変更申請書に加えて逸脱報告書・品質情報報告書・年次照査の自動生成にも対応しました。
QAI Generatorを導入することで、データインテグリティ担保のための文書作成にかかる時間を削減し、皆様がより本質的な品質保証業務に専念できる環境づくりをサポートします。
まとめ
本記事では、製薬業界におけるデータインテグリティの重要性について解説しました。
- データインテグリティとは: データの「完全性・一貫性・正確性」を保証することであり、規制当局も監視を強めています。
- ALCOA原則の遵守: データの信頼性を守るための9つの要件(ALCOA+CCEA)を理解し、実践する必要があります。
- 業務効率化の鍵はAI: 厳格化する規制に対応しながら負担を減らすには、AIツールの活用が有効です。
データインテグリティへの対応は、企業の信頼と患者様の安全を守るための最優先事項です。しかし、すべてをマンパワーで解決する必要はありません。
弊社が提供する「QAI Generator」は、GMP文書業務の効率化を強力にバックアップいたします。データインテグリティ対応にお悩みの方は、ぜひ一度、資料請求にて詳細をご覧ください。
関連記事はこちら


